



İçindekiler
Prion Nedir?
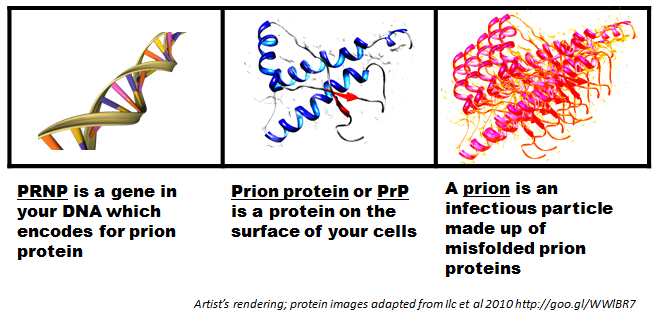
BSE, Scrapie ve Creutzfeldt-Jakob hastalığı gibi beyin hastalıklarının nedeni olduğu düşünülen bir protein parçacığıdır. Diğer isimleri CD230 ve PrP olan bu proteini PRNP geni kodlar. Nükleik asit içermemeleri dolayısıyla prionlar diğer tüm patojen türlerinden (bakteri, mantar, virüs vb.) farklıdır ve diğer patojen türlerini yok etmek için kullanılan hiçbir nükleik asit parçalama yöntemi prionlarda işe yaramaz yani yıkıma(destruction) yok edilme karşı oldukça dirençlidirler. Dahası, prionlar beden tarafından kodlanan normal bir proteinin anormal şekilde katlanmış versiyonu olduğundan, herhangi bir bağışıklık tepkisi de üretmezler. Prion tek başına mikroskobik olarak görünmez, virüslerden bile daha küçük olan prionlar, ancak birikip kümelendiklerinde elektron mikroskobu altında görülebilir. Bu proteinin kesin işlevi bilinmemekte fakat araştırmacılar, bakırın hücrelere taşınması, nöronların yaralanmaya karşı korunması (nöroproteksiyon) ve nöronlar arasındaki iletişim gibi görevleri olduğu hakkında çeşitli hipotez ve teoriler üretiyor.
Prion Hastalığı
Prion hastalığı, insanlarda ve hayvanlarda sinir sistemini etkileyen bir grup durumu temsil eder. İnsanlarda bu koşullar beyin işlevini bozarak hafıza, kişilik ve davranışta değişikliklere neden olur; entelektüel işlevde bir düşüş (demans) ve anormal hareketler, özellikle hareketleri koordine etmede zorluk (ataksi) belli başlı olanlardır. Prion hastalığının belirti ve semptomları tipik olarak yetişkinlikte başlar ve zamanla kötüleşerek birkaç aydan birkaç yıla kadar ölüme yol açar.
Prion Hastalığının Diğer İsimleri
- Kalıtsal insan bulaşıcı süngerimsi ensefalopatiler
- Prion protein hastalıkları
- Prion ile ilişkili bozukluklar
- Prion kaynaklı bozukluklar
- Bulaşıcı demanslar
- Bulaşıcı süngerimsi ensefalopatiler
- TSEs
Sıklık
Prion hastalığının görülme sıklığı kesin bilinmemekle birlikte, araştırmalar bu hastalık grubunun her yıl dünya çapında milyonda bir kişiyi etkilediğini göstermektedir. Amerika Birleşik Devletleri’nde yılda yaklaşık 350 yeni vaka rapor edilmektedir.
Prion Hastalığının Aşamaları ve Türleri
Prion hastalığı vakalarının yüzde 10-15’i PRNP genindeki mutasyonlardan kaynaklanmaktadır. Bu yüzde grubundaki tür, kalıtsal olabildiği için ailesel olarak sınıflandırılır. Prion hastalığının ailesel formları, otozomal dominant bir modelde kalıtılır; bu, bozukluğa neden olmak için her hücrede değiştirilmiş PRNP geninin bir kopyasının yeterli olduğu anlamına gelir.
Birbirleriyle örtüşen semptomları olan ailesel prion hastalıkları arasında ailesel Creutzfeldt-Jakob hastalığı (CJD), Gerstmann-Sträussler-Scheinker sendromu (GSS) ve ölümcül ailesel uykusuzluk (FFI) bulunur. Prion hastalığının ailesel formlarında, PRNP gen mutasyonları, genin bir kopyasından PrPSc olarak bilinen anormal şekilli bir proteinin üretilmesine neden olur. PrPSc normal protein olan PrPC’ye(PrP’nin glikoprotein gibi hücresel formudur) bağlanabilir ve bunun PrPSc’ye dönüşümünü teşvik edebilir. Anormal protein beyinde birikerek nöronlara zarar veren kümeler oluşturur. Bu hücrelerin kaybı, beyinde, prion hastalığının semptomlarına yol açan mikroskobik sünger benzeri delikler (vakuoller) meydana getirir.
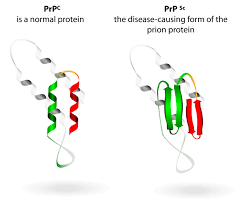
Prion hastalığı vakalarının diğer yüzde 85 ila 90’ı, sporadik, edinilmiş ve iyatrojenik olarak sınıflandırılır. Sporadik prion hastalığı olan kişilerin ailesinde bu hastalık ve PRNP geninde tanımlanmış bir mutasyon yoktur. Sporadik hastalık, PrPC kendiliğinden ve bilinmeyen nedenlerle PrPSc’ye dönüştürüldüğünde ortaya çıkar. Prion hastalığının sporadik formları arasında sporadik Creutzfeldt-Jakob hastalığı (sCJD), sporadik ölümcül uykusuzluk (sFI) ve değişken olarak proteaza duyarlı prionopati (VPSPr) bulunur.
Nadiren, prion hastalığı, tıbbi bir prosedür sırasında yanlışlıkla PrPSc ile kontamine olmuş dokulara maruz kalma ile bulaşabilir. Tüm vakaların yüzde 1 ila 2’sini oluşturan bu tip prion hastalığı, iyatrojenik olarak sınıflandırılır.
Edinilmiş prion hastalığı, bir dış kaynaktan PrPSc’ye maruz kalmanın sonucudur. Örneğin, varyant Creutzfeldt-Jakob hastalığı (vCJD), prion hastalığı olan sığırlardan elde edilen PrPSc içeren sığır eti ürünlerini yemekten kaynaklanan edinilmiş bir prion hastalığı türüdür. İneklerde, hastalığın bu formu sığır süngerimsi ensefalopati (BSE) veya yaygın adı “deli dana hastalığı” olarak bilinir. Bir başka örnek, Papua Yeni Gine’deki Güney Fore popülasyonunda tespit edilen Kuru’dur. Bozukluk, yamyamlık cenaze törenleri sırasında bireyler başka bir etkilenmiş insan dokusunu yediğinde bulaşmıştır.
Kuru ve varyant Creutzfeldt-Jakob hastalığı dahil olmak üzere prion hastalığının sporadik, edinilmiş ve iyatrojenik formları kalıtsal değildir.
Bazı insanlarda, prion hastalığının ailesel formlarına, bir ebeveynin üreme hücrelerinin (yumurta veya sperm) oluşumu veya erken embriyonik gelişim sırasında gende meydana gelen yeni bir mutasyon neden olur. Bu insanların etkilenmiş bir ebeveyni olmamasına rağmen bu kişiler genetik değişikliği çocuklarına aktarabilirler.
Kaynaklar:
- https://labtestsonline.org.tr/glossary/prion
- https://medlineplus.gov/genetics/condition/prion-disease/#causes
- https://bilimfili.com/prion-arastirmalari-icin-yeni-bir-yontem-gelistirildi
Görsel Kaynak: https://cjdfoundation.org/about-cjd
Editör: Meryem Melisa KAR
