
İçindekiler
Tanım
Sly sendromu olarak da bilinen mukopolisakkaridoz tip VII (MPS VII), birçok doku ve organı etkileyen, ilerleyen bir durumdur. MPS VII’nin şiddeti, etkilenen bireyler arasında büyük ölçüde değişir.
En şiddetli MPS VII vakaları, doğumdan önce vücutta fazla sıvının birikmesi durumu olan hidrops fetalis ile karakterize edilir. Hidrops fetalisli birçok bebek, ölü doğar veya doğumdan hemen sonra vefat eder. MPS VII’li diğer kişiler, genellikle bu durumun belirti ve semptomları göstermeye erken çocukluk döneminde başlar. MPS VII’nin özellikleri, geniş kafa (makrosefali), beyinde sıvı birikmesi (hidrosefali), kaba, kalın olarak tanımlanan farklı görünen yüz özellikleri ve büyük dili (makroglosi) içerir.
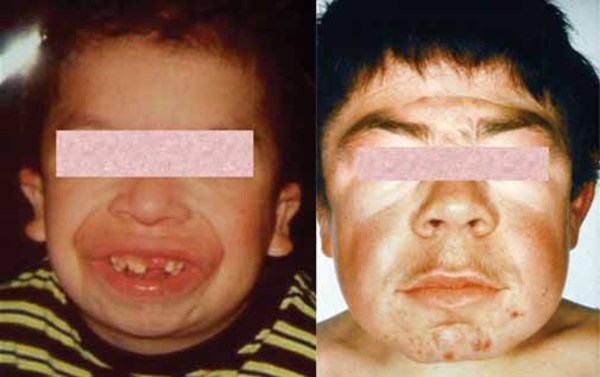
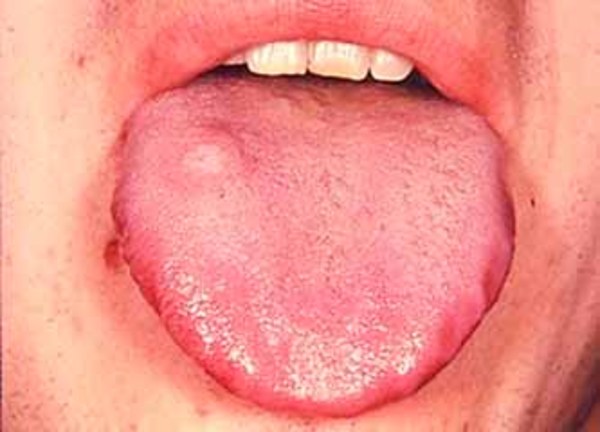
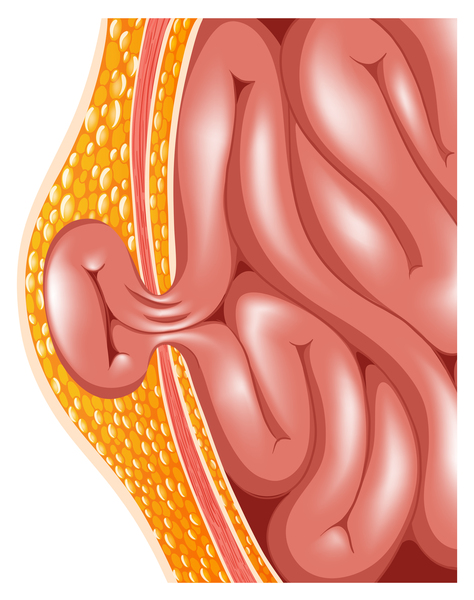
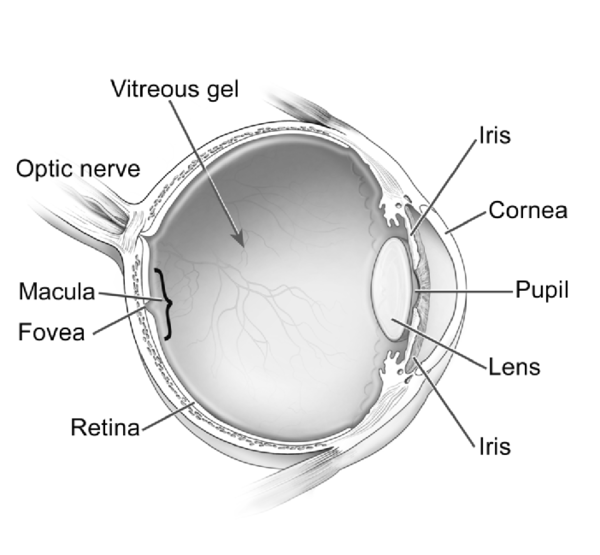
Etkilenen bireylerde ayrıca, genişlemiş karaciğer ve dalak (hepatosplenomegali), kalp kapakçık anormallikleri ve göbek deliği (göbek fıtığı) veya alt karın (kasık fıtığı) etrafında yumuşak kese geliştirir. MPS VII’li bazı kişilerde havayolu daralabilir; bu da sıklıkla üst solunum yolu enfeksiyonlarına ve uyku sırasında nefes almada kısa duraklamalara (uyku apnesi) neden olur. Gözün şeffaf örtüsünün (kornea) bulanıklaşması, önemli görme kaybına neden olabilir. MPS VII’li kişiler ayrıca, tekrarlayan kulak enfeksiyonları ve duyma kaybına sahiptir. Bu duruma sahip bazı bireylerde zekâ etkilenmemesine rağmen, etkilenen bireyler, gelişimsel gecikme ve ilerleyen zihinsel engelliliğe sahiptir.
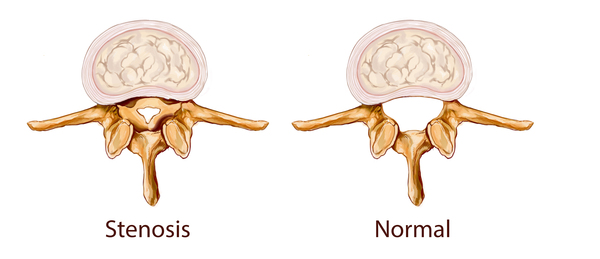
MPS VII, kısa boy ve hareketi etkileyen eklem bozukluklarını (kontraktürler) içeren, yaşla beraber daha belirgin olan çeşitli iskelet anormalliklerine sebep olur. Bu duruma sahip bireyler ayrıca, x-ray de görülen çoklu iskelet anormalliklerini ifade eden dizostoz multiplekse sahip olabilir. Karpal tünel sendromu (sinir başı sendromu), MPS VII’li birçok çocukta gelişir ve elde ve parmaklarda uyuşma, karıncalanma ve zayıflık ile karakterize edilir. MPS VII’li bireyler, boyunda omuriliği sıkıştırabilecek ve hasar verebilecek omurilik (spinal) kanalının daralmasını (spinal stenoz) geliştirebilir.
MPS VII’li bireylerde yaşam beklentisi, semptomların şiddetine bağlıdır. Bazı etkilenen bireyler, bebeklik döneminde hayatta kalmaz. Diğerleri, ergenlik ve yetişkinliğe kadar yaşayabilir. Kalp hastalıkları ve hava yolu tıkanıklığı, MPS VII’li kişilerin ana ölüm nedenleridir.
Sıklık
250.000 yeni doğandan 1’inde oluştuğu tahmin edilmesine rağmen, MPS VII’nin kesin görülme oranı bilinmemektedir. En nadir görülen mukopolisakkaridoz türlerinden biridir.
Nedenler
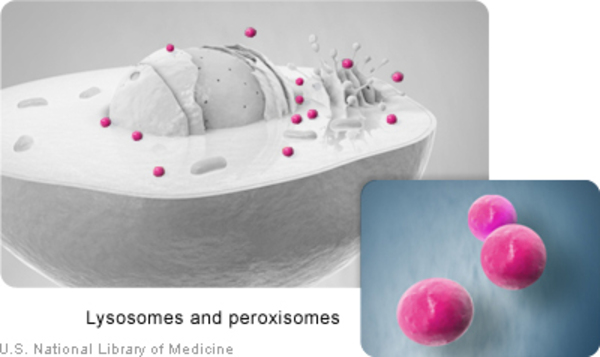
GUSB genindeki mutasyonlar, MPS VII’e neden olur. Bu gen, glikozaminoglikanlar (GAG’lar) olarak adlandırılan, büyük şeker moleküllerinin yıkımında yer alan beta-glukuronidazın (β-glukuronidaz) üretimi için bilgi sağlar. GAG’lar, aslında bu durumun adını aldığı mukopolisakkaridoz olarak adlandırılırdı. GUSB genindeki mutasyonlar, β-glukuronidaz fonksiyonunu azaltır veya tamamıyla yok eder. β-glukuronidazın kıtlığı (yetersizliği), hücrelerde, özellikle lizozomların içinde, GAG’ların birikmesine neden olur. Lizozomlar, hücrede, farklı molekül türlerini geri dönüştüren ve sindiren elemanlardır. Moleküllerin lizozomların içinde birikmesisine neden olan MPS VII gibi durumlar, lizozomal depo rahatsızlıkları olarak adlandırılır. GAG’ların birikimi, lizozom boyutunu arttırır; bu da, bu rahatsızlıkta birçok doku ve organın genişlemesini açıklar. Araştırmacılar, GAG’ların ayrıca lizozomların içindeki diğer proteinlerin işlevine engel olabileceğine ve hücrelerin birçok normal işlevini sekteye uğratabileceğine inanıyorlar.
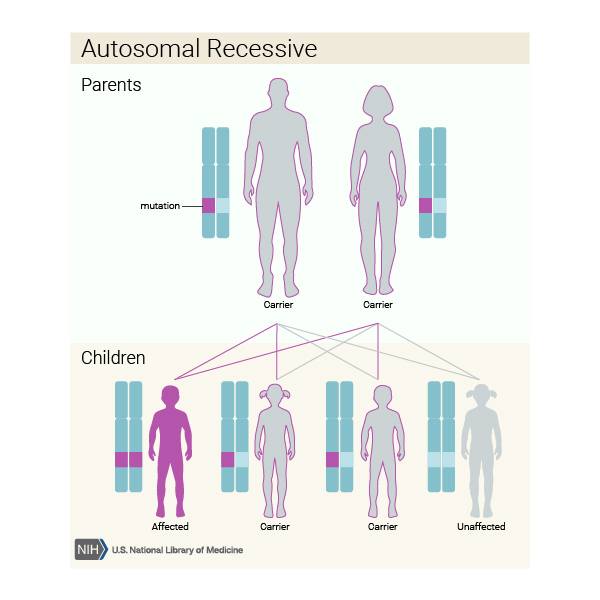
Kalıtım Modeli
Bu durum otozomal çekinik modelde kalıtılır. Bu da, her hücredeki iki kopyanın mutasyona sahip olduğu anlamına gelir. Otozomal çekinik duruma sahip bireyin ebeveynlerinin her biri mutasyonlu genin bir kopyasını taşır ama tipik olarak bu durumun belirti ve semptomlarını göstermezler.
Bu Hastalığın Diğer İsimleri
- β-glukuronidaz eksikliği
- GUSB eksikliği
- MPS VII
- MPS7
- Mukopolisakkaridoz 7
- Mukopolisakkaridoz VII
- Sly sendromu
Kaynak: https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-vii/
Görsel Kaynak: https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-vii/
Editör: Seray YETKİN
