




Tanım
SMA yani tam ismiyle spinal müsküler atrofi, omurilikte bulunan alfa motor nöronların dejenerasyonu ve kaybı ile karakterize, otozomal resesif olarak aktarılan nöromüsküler bir hastalıktır. Kalıtsal olarak aktarılan SMA, nadir hastalıklar kategorisindedir. Henüz kesin bir tedavisi olmamakla birlikte hastalığın moleküler genetiğinin anlaşılması yapılan çalışmalara katkı sağlamış, çok sayıda potansiyel terapötik yaklaşımın geliştirilmesine yol açmıştır. İlk olarak Guido Werdnig tarafından tanımlanan SMA, 1890’larda Johan Hoffmann tarafından 7 ayrı vakada daha tespit edilmiştir. SMA görülme insidansı (sıklığı) canlı doğumlarda 1: 11.000 olarak bildirilmiştir (1-4).
SMA’ya Dair Konsensus ve Klinik Sınıflandırma
Uluslararası SMA için Standart Bakım Komitesi, 2005 yılında SMA alanında klinikteki durumu araştırmak ve hastaların klinik bakımında kullanılmak üzere kılavuz niteliği taşıyacak bir fikir birliğine ulaşmak amacıyla kuruldu. 60’dan fazla araştırmacı; teşhis, bakım, beslenme, ortopedi, rehabilitasyon ve diğer birçok alanda çalışmalar yaptı. Grupların çalışmalarının sonuçları yayınlar, toplantılar, konferanslar vb. ile diğer gruplarla paylaşıldı ve 2007 yılında bir konsensüs yayınlandı. Yayınlanan konsensüs oturmayan, oturabilen ve yürüyebilen grupları içeriyordu. SMA için kullanılan teşhis süreci bu bildiriden temel almakla birlikte, daha sonra yapılan çalışmalarla SMA tipleri detaylandırılmış ve genetik arka plana dair bilgiler artmıştır. Tanı sürecini, ailesel vakalar dışında klinik belirtilerin yönlendirdiği bilinmektedir (5,6).
SMA’da motor nöronların kaybına bağlı olarak en çok görülen klinik özellikler; kaslarda güçsüzlük ve atrofidir. Hastalığın şiddet spektrumu geniştir. Yetişkinlikte hafif proksimal uzuv zayıflığı görülebildiği gibi, yenidoğanlarda solunum yetmezliği ile şiddetli genel zayıflık görülebilir. Alt ekstremiteler, üst ekstremitelere göre daha fazla etkilenmektedir (3, 7).
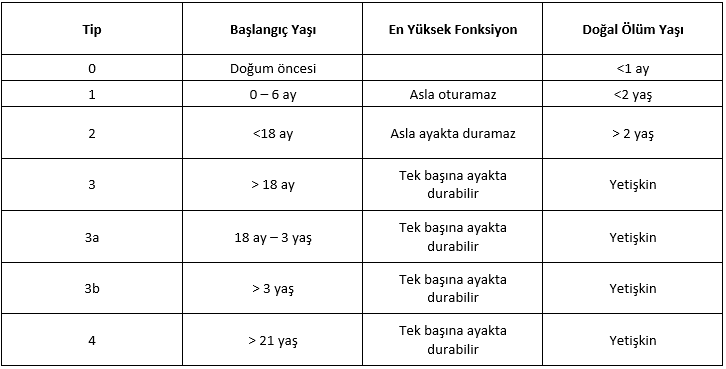
SMA tipleri başlangıçta oturma, ayakta durma gibi en yüksek motor fonksiyon seviyesine ve başlangıç yaşına göre SMA tip 1,2 ve 3 olmak üzere 3’e ayrılmış; daha sonra sınıflandırmaya SMA tip 0 ve 4 eklenmiştir (Tablo 1) (3).
SMA’nın Moleküler Genetiği ve Tanı Yöntemleri
SMA, 5q kromozomu üzerinde bulunan SMN1 geninin delesyona ve mutasyona uğraması sonucu oluşan otozamal resesif bir hastalıktır. Hastaların ~%95’inde SMN1’nin homozigot delesyonu/mutasyonu (ekzon 7 ve ekzon 8’de), ~%2’sinde SMN alellerinden birinde de novo (geri dönüşümsüz) delesyonlar ve ~%3 – %4’ünde diğer mutasyonlar (nokta mutasyonları) vardır. SMN1 gen ürünü olan SMN proteini motor nöron gelişimi için büyük önem taşır (8, 9).
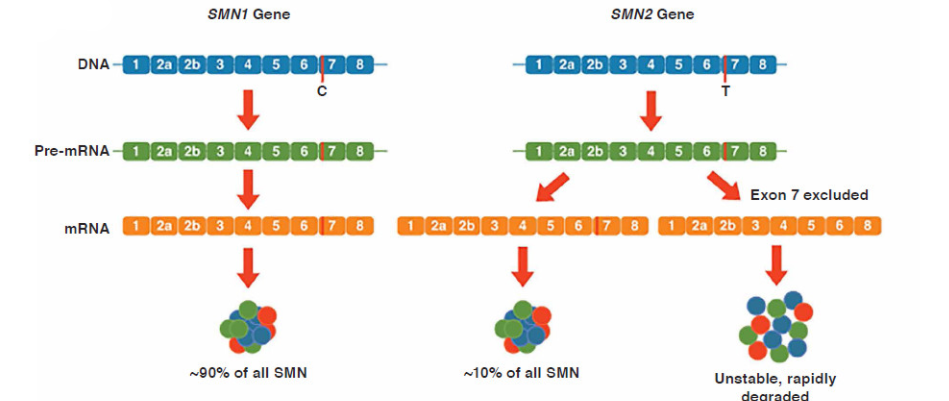
SMN1’in bulunduğu kromozom üzerinde SMN1 proteinine çok benzer yapıda bulunan SMN2 proteini vardır. SMN2’nin SMN1’den tek farkı; C nükleotidinden T nükleotidine geçişte translasyonel olarak sessiz olmasıdır. Bu durum SMN2 transkriptlerinin birleştirilmesinde ekzon 7’nin atlanmasına ve hızla bozulan kararsız bir proteinin oluşmasına sebep olur. mRNA’ların sadece çok küçük bir kısmı tüm ekzonu içeren stabil SMN proteini kodlar (Şekil 1). SMN proteinin tamamen yokluğu ölümcüldür. SMA’lı bireylerde en az bir tane SMN2 kopyası vardır. Kopya sayısı hastalıkla ilişkili olmakla birlikte daha hafif fenotiplere ilgili daha fazla sayıda (SMA tip 1 için 1-2 kopya varken, tip 3 için 3-4) kopya vardır (8-10).
SMA tanısında tipik bir vakada ilk basamak, SMN1/SMN2’nin kantitatif analizinin yapıldığı genetik testlerdir. SMN1’in 0 kopyası olması durumunda SMA teşhisi direkt konulur, tek kopya mevcutsa olası diğer mutasyonların araştırılması için SMN1 geni dizilenir. Her iki tam kopya mevcutsa SMA olası olmamakla birlikte, tipik bir fenotip veya akrabalık durumu varsa SMN1 geni dizilenmelidir. Kopya sayıların hastalığın şiddetini etkilediği bilinse de tam bir korelasyon göstermediği bilinmektedir. Bu nedenle bir kişide hastalığın ciddiyeti kopya sayısı kullanılarak tahmin edilemez (5, 11).
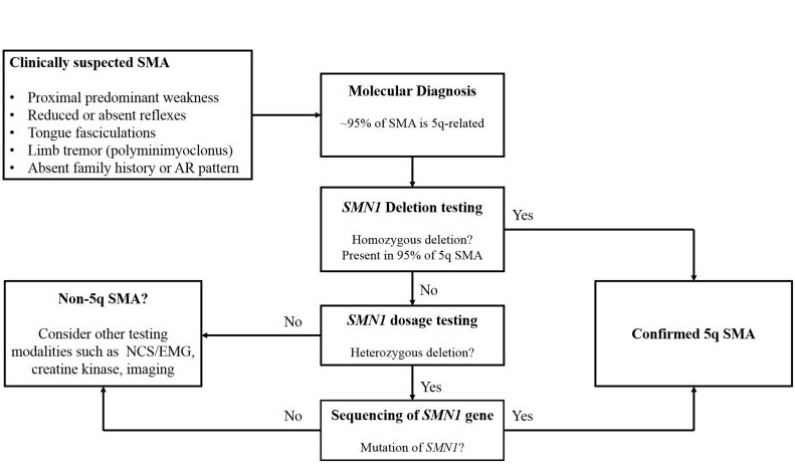
SMA için taşıyıcılık testleri de mevcuttur. Taşıyıcılık frekansı çalışılan popülasyona bağlı olmakla birlikte 1/40-1/60’dır. SMN1 dozaj testleri ile taşıyıcıların büyük bir kısmının (%95) tespiti için yeterlidir. Heterozigot durumda olanların tespiti içinse RT-PCR ya da Q-PCR yöntemleri kullanılarak yapılan testler geliştirilmiştir. SMA’nın diğer otozomal resesif hastalıklara kıyasla yüksek taşıyıcılık oranına sahip olmasının sebebi; SMN1/SMN2 lokusu çevresinde çok fazla sayıda tekrarlayan sekans bulunması ve bu sebeple bölgede gerçekleşen de novo mutasyon oranının artması olarak gösterilebilir (5, 12).
SMA’nın erken gebelikte ve yenidoğanlarda tespiti için de çalışmalar yapılmaktadır. Yenidoğanlarda görüntüleme çalışmaları kimi ülkelerde halihazırda uygulamaya konmuş, bazı ülkelerse uygulanabilirlik ve ekonomik süreci değerlendirilmektedir. Yenidoğan taraması asemptomatik dönemde tanı koymak için güzel bir seçenek olsa da SMN1 geninde nokta mutasyonu olan hastaların tespiti yapılamamaktadır. Erken gebelikte yapılan testler de yine erken teşhise olanak tanımasına karşın, çalışmalar invazivdir ve anne/bebek için ciddi riskler taşır. Maternal kandan fetal DNA’nın izolasyonu ile yapılan çalışmalarda mevcuttur fakat teknik sınırlamaların yanı sıra yüksek maliyet ve özel laboratuvar gereksinimi ülke çapında uygulama yapılmasını zorlaştıran etmenlerdir (13).
Kısaltmalar:
- SMA: Spinal müsküler atrofi
- SMN: Yaşamsal motor nöron (survival motor neuron)
- m RNA: Mesajcı ribonükleik asit
- DNA: Deoksiribonükleik asit
- C: Sitozin
- T: Timin
- RT-PCR: Gerçek zamanlı polimeraz zincir reaksiyonu
- Q-PCR: Yarışmalı polimeraz zincir reaksiyonu
Kaynaklar:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3231874/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3860273/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4628728/
- https://pubmed.ncbi.nlm.nih.gov/21811307/
- https://pubmed.ncbi.nlm.nih.gov/29290580/
- https://pubmed.ncbi.nlm.nih.gov/17761659/
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4293319/
- https://pubmed.ncbi.nlm.nih.gov/30582825/
- https://pubmed.ncbi.nlm.nih.gov/29478602/
- https://pubmed.ncbi.nlm.nih.gov/25413156/
- https://pubmed.ncbi.nlm.nih.gov/25346245/
- https://pubmed.ncbi.nlm.nih.gov/20057317/
- https://pubmed.ncbi.nlm.nih.gov/30656198/
Görsek Kaynak: http://europepmc.org/articles/PMC4628728/figure/F3/
Editör: Seray YETKİN

1 Yorum