

İçindekiler
COL1A1 Geni: Kollajen Tip 1 Alfa 1 Zinciri
Normal Fonksiyonu
COL1A1 geni, tip1 kollajen olarak adlandırılan büyük bir molekülün bir kısmını yapmak için emirler verir. Kollajenler, kıkırdak, kemik, tendon, cilt ve gözün beyaz kısmı (sklera) dahil olmak üzere vücuttaki birçok dokuyu güçlendiren ve destekleyen protein ailesidir. Tip 1 kollajen, insan vücudunda en çok bulunan kollajen formudur.
Pro-α1(I) zinciri olarak adlandırılan tip 1 kollajenin bir bileşeni, COL1A1 geninden üretilir. Kollajenler, her biri üç zincirden oluşan, halat benzeri prokollajen molekülleri olarak başlar. Tıp 1 kallojen iki pro-α1(I) zincirinden ve bir pro-α2(I) zincirinden (COL1A2 geninden üretilen) oluşur.
Üç iplikçikli prokollajen molekülleri, olgun kollajen oluşturmak için hücrenin içindeki ve dışındaki bir dizi adımda enzimler tarafından işlenir. Kollajen molekülleri daha sonra kendilerini hücreler arasındaki boşluklarda birbirleriyle stabil etkileşimler (çapraz bağlantılar) oluşturan uzun, ince fibrillere yerleştirirler. Çapraz bağlantılar, çok güçlü tip 1 kollajen liflerinin oluşumu ile sonuçlanır.
Genetik Değişikliklerle İlgili Sağlık Durumları
Caffey Hastalığı
COL1A1 genindeki belirli bir mutasyon, yaygın olarak Caffey hastalığı olarak bilinen İnfantil Kortikal Hiperastoz’a neden olur. Caffey Hastalığı’nın belirtileri ve semptomları genellikle bir bebek beş aylık olduğu zaman ortaya çıkar. Bu durum yumuşak dokuların şişmesi (örneğin kaslar), ağrı ve aşırı yeni kemik oluşumu (hiperostoz) ile karakterizedir. Kemik anormallikleri esas olarak çene kemiğini, köprücük kemiğini (klavikulaları) ve kol ve bacaklardaki uzun kemiklerin şaftlarını (diyaframlar) etkiler. Bilinmeyen nedenlerden dolayı, Caffey hastalığı ile ilişkili ağrı ve şişlik tipik olarak birkaç ay içinde kaybolur. Eski kemik dokusunu yeni kemikle değiştiren kemik yeniden modelleme adı verilen normal bir işlemle, fazla kemik genellikle vücut tarafından yeniden emilir ve 2 yaşına kadar x-ışını görüntülerinde saptanamaz.
Bu duruma neden olan mutasyon, her hücrede COL1A1 geninin bir kopyasında meydana gelir. 836 protein konumundaki amino asit sistein ile amino asit arginin (Arg 836 of R836C olarak yazılır) yer değiştirmesi ile tek bir protein yapıtaşını (amino asit) değiştirir. Bu mutasyon, boyut ve şekil olarak değişken olan tip I kollajen fibrillerinin üretilmesine neden olur, ancak bu değişikliklerin Caffey Hastalığı’nın belirtilerine ve semptomlarına nasıl yol açtığı bilinmemektedir.
Ehlers-Danlos Sendromu
COL1A1 genindeki mutasyonların, cildi, kemikleri, kan damarlarını ve diğer birçok organ ve dokuyu destekleyen bağ dokularını etkileyen bir grup bozukluk olan çeşitli Ehlers-Danlos sendromu formlarına neden olduğu bulunmuştur. Bu mutasyonlar, her hücrede COL1A1 geninin bir kopyasından meydana gelir.
COL1A1 genindeki en az beş mutasyon, alışılmadık derecede geniş eklem hareket aralığı (hipermobilite) ve doğumda her iki kalçanın yerinden çıkmasıyla karakterize olan, Artrokalazi tipi Ehlers-Danlos Sendromu ile sonuçlanabilir. Hastalığın bu formuna neden olan genetik değişikler, kritik bir segmenti eksik olan, pro-α1(I) zincirinin üretilmesine neden olur. Bu segmentin eksikliği, pro-α1(I) zincirlerinin olgun tip 1 kollajen moleküllerine birleştirilmesine ve işlenmesine engel olur. Deri, kemik ve tendonlar gibi tip 1 kollajen bakımından zengin dokular, bu değişiklikten en çok etkilenir.
COL1A1 gen mutasyonları, ayrıca Ehlers-Danlos Sendromu’nun klasik ve vasküler tiplerinin nadir bir nedenidir. (Çoğu vakada, bu tipler diğer genlerdeki mutasyonlardan kaynaklanır.) Klasik tip yumuşak, oldukça esnek (elastik) ve kırılgan olan cilt; anormal yara izi ve eklem hipermobilitesi ile karakterize edilir. Ek olarak, bir COL1A1 gen mutasyonundan kaynaklanan klasik Ehlers-Danlos sendromu olan bireyler, erişkinlikte büyük arterlerin yırtılmasına (raptürüne) meyillidir. Vasküler tip, kan damarlarının, bağırsakların ve diğer organların yırtılması ile ilişkilidir. Hem klasik hem de vasküler Ehlers-Danlos Sendromu ile ilişkili bir COL1A1 gen mutasyonu, amino asit argininini pro-a1 (I) zincirinde 312 pozisyonunda (Arg312Cys veya R312C olarak yazılır) amino asit sistein ile değiştirir. Değiştirilmiş pro-α1(I) zinciri, tip 1 kollajen fibrillerinin yapısını bozarak, hücre içinde yakalayarak, diğer kollajen yapıcı proteinlere müdahale eder. Kollajenlerdeki bu değişiklikler, kan damarı ve organ yırtılma riskini ve Ehlers-Danlos Sendromu’nun klasik ve vasküler tiplerinde ortaya çıkabilecek diğer anormallikleri arttırır.
Osteogenez İmperfektası
COL1A1 genindeki mutasyonların neden olduğu en sık görülen hastalık Osteogenez İmperfektası’dır. Bu rahatsızlığa sahip olan kişiler, genellikle hafif travmadan veya belirgin bir nedeni olmayan, kolayca kırılan kemiklere sahiptir. Ek olarak, etkilenen bireyler, gözün genellikle beyaz olan kısmında (sklera) mavi veya gri bir renk tonu, kısa boy, işitme kaybı, solunum problemleri ve Dentinogenez İmperfekta adı verilen diş gelişimi bozukluğuna sahip olabilir. Osteogenez İmperfekta’ya neden olan yüzlerce COL1A1 gen mutasyonu tanımlanmıştır. Bu hastalığın en hafıf formu olan Osteogenez İmperfecta tip I’den sorumlu mutasyonların çoğu, pro-α1(I) zincirlerinin üretimini azaltır. Daha az sayıda pro-α1(I) zinciri mevcut olduğunda, hücreler normal tip 1 kollajen miktarının sadece yarısını yapabilirler. Bu kritik proteinin eksikliği, kemik kırılganlığının ve Osteogenez İmperfecta tip 1’ in diğer karakteristik özelliklerinin altındadır.
COL1A1 genindeki çeşitli mutasyonlar, tip 1, 2, 3 ve 4 dahil olmak üzere daha ciddi osteogenez İmperfekta formlarına neden olur. Bu mutasyonların bazıları, DNA segmentlerini COL1A1 geninden siler, bu da anormal olarak kısaltılmış pro-α1(I) zinciri ile sonuçlanır. Diğer genetik değişiklikler, pro-α1(I) zincirinde, genellikle amino asit glisini farklı bir amino asitle değiştirerek, amino asitlerin yapısını değiştirir. Bazı durumlarda, amino asit ikameleri, kollajen moleküllerin birleşmesini engelleyen protein zincirinin bir ucunu (C-terminus olarak adlandırılır) değiştirir. Bu COL1A1 gen mutasyonları, tip 1 kollajenin anormal versiyonlarının üretilmesine yol açar. Bu anormal kollajen, gelişmekte olan kemiklere ve diğer bağ dokularına dahil edildiğinde, şiddetli Osteogenezis İmperfekta formlarıyla ilişkili ciddi sağlık sorunlarına neden olur.
Karpal Tünel Sendromu
Dermatofibrosarkom Protuberans
Derinin derin tabakalarında bir tümöre neden olan nadir bir kanser türü olan Dermatofibrosarkom Protuberans , COL1A1 genini içeren kalıtsal olmayan (somatik) bir mutasyon ile karakterizedir. Somatik mutasyonlar bir insanın yaşamı boyunca kazanılır ve sadece belirli hücrelerde bulunur, bu durumda derideki hücreler kanserden kaynaklanır. Dermatofibrosarkom Protuberans, 17 ve 22 kromozomları arasında genetik materyalin yeniden düzenlenmesi (translokasyon) ile ilişkilidir. T(17; 22) olarak yazılan bu translokasyon, COL1A1 geninin bir kısmını, kromozom 17 üzerinde, bir genin bir kısmı ile PDGFB adı verilen kromozom 22 üzerinde birleştirir. Bu translokasyon, normal doğrusal şekil veya dairesel olabilen bir veya daha fazla kromozom üzerinde bulunur.
Kaynaşmış COL1A1-PDGFB geni, araştırmacıların sonuç olarak aktif PDGFB proteini gibi işlev gördüğüne inanan, bileşik (füzyon) bir proteinin üretim emrini verir. Translokasyonda PDGFB geni, DNA’sının aktivitesini sınırlayan kısmını kaybeder ve COL1A1-PDGFB füzyon proteininin üretimi COL1A1 gen dizileri tarafından kontrol edilir. Sonuç olarak, gen füzyonu normalden daha fazla miktarda aktif PDGFB proteini üretimine yol açar. Aktif PDGFB proteini üretimine yol açar. Aktif PDGFB proteini, hücre büyümesi ve bölünmesi (çoğalması) ve olgunlaşması (farklılaşması) için sinyal verir. Fazla PDGFB proteini anormal bir şekilde hücreleri çoğalmaya ve farklılaşmaya teşvik ederek Dermatofibrosarkom Protuberası’nda tümör oluşumuna yol açar.
İntervertebral Disk Hastalığı
Diğer Bozukluklar
Bazı COL1A1 mutasyonlarına sahip kişiler, hem Osteogenez İmperfekta hem de Ehlers-Danlos Sendromu’nun (yukarıda tarif edilen) belirtilerini ve semptomlarını sergilerler. Bu mutasyonlar genellikle amino asit glisini, pro-a1 (I) zincirindeki farklı bir amino asit ile değiştirir, bu da pro-a1 (I) zincirlerinin olgun tip I kollajen moleküllerine birleştirilmesini ve işlenmesini engeller. Ortaya çıkan anormal tip 1 kollajen fibrilleri bağ dokusunu zayıflatarak bu iki durumla ilişkili belirtilere ve semptomlara neden olur.
COL1A1 genindeki (bir polimorfizm olarak adlandırılan) yaygın bir değişiklik, osteoporoz gelişme riskini arttırıyor gibi görünmektedir. Osteoporoz, kemikleri aşamalı olarak daha kırılgan yapan ve kırılmaya eğilimli hale getiren bir durumdur. COL1A1 geninin kontrol (düzenleyici) bir bölgesinde meydana gelen bu polimorfizm, muhtemelen tip 1 kollajen üretimini etkiler, ancak molekülün yapısını etkilemez. Birçok çalışma, bu genetik değişime sahip kadınların, osteoporoz belirtilerine, özellikle düşük kemik yoğunluğuna ve kemik kırıklarına sahip olma olasılığının, değişikliği olmayan kadınlardan daha olası olduğunu göstermiştir. Bu varyasyon, osteoporoz riskini arttırabilecek birçok faktörden sadece biridir.
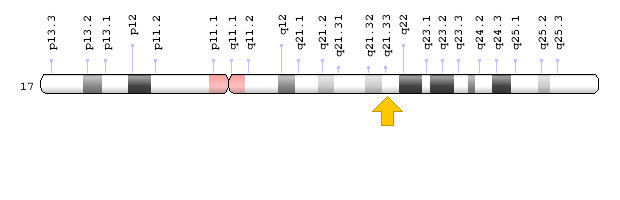
Kromozomal Konum
- Sitogenetik Konum: 17q21.33, 21.33 konumundaki kromozom 17‘nin uzun (q) kolu
- Moleküler Konum: Kromozom 17 üzerindeki 50,184,096 – 50,201,649 baz çiftleri
Genin Diğer İsimleri
- Alfa 1 Tip 1 Kollajen Preproprotein
- CO1A1_İNSAN
- COL1A1 Protein
- Kollajen 1, Alfa-1 Polipeptit
- Cilt, Tendon ve Kemik Kollajeni, Alfa-1 Zinciri
- Kollajen Tip 1 Alfa 1
- Kollajen, Tip 1, Alfa 1
- Tip 1 Kollajen Alfa 1
Kaynak: https://ghr.nlm.nih.gov/gene/COL1A1
Görsel Kaynak: https://www.rcsb.org/3d-view/ngl/1t60
Editör: Meryem Melisa KAR
