
İçindekiler
Tanım
Camurati-Engelmann Hastalığı, kollarda, bacaklarda ve kafatasında anormal derecede kalın kemikler (hiperostoz) ile karakterize edilen bir iskelet hastalığıdır.
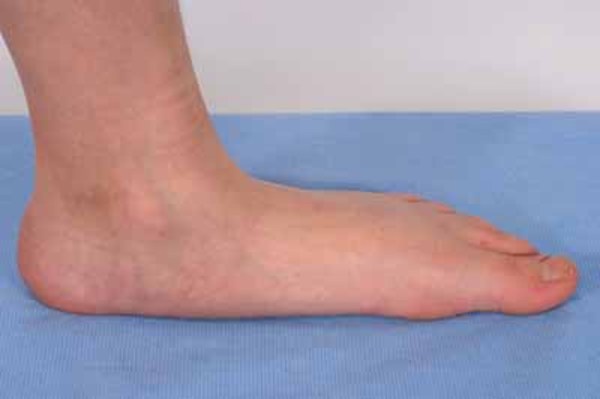
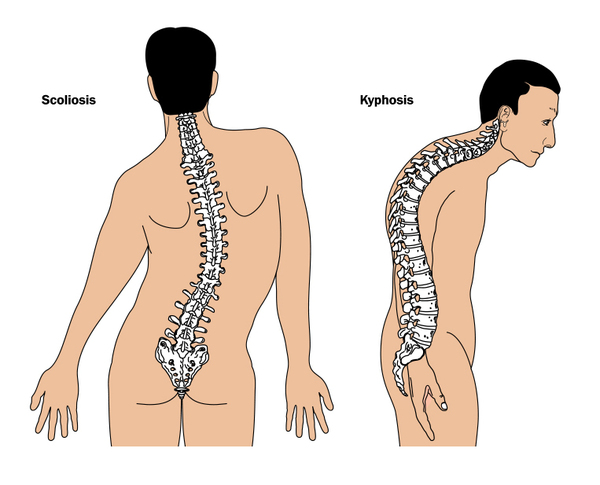
Kalın uzuv kemikleri, kollarda ve bacaklarda kemik ağrısına ve kas güçsüzlüğüne sebep olabilir ve Camurati-Engelmann Hastalığı olan bireylerin çabuk yorulmasına neden olabilir. Kemik ağrısı hafif ile şiddetli arasında değişebilir ve stres, aktivite ya da soğuk havalarda artabilir. Bacak güçsüzlüğü, oturur pozisyondan ayağa kalkmayı zorlaştırabilir ve etkilenen bazı bireylerde, yalpalama ya da kararsız yürüyüş gelişir. Buna ek olarak, uzuv anormallikleri eklem deformasyonlarını (kontraktürler), çarpık bacaklığı (Görsel 1) ve düztabanlığı (pes planus) (Görsel 2) içerir. Uzuvlarda şişme ve kızarıklık ve omurganın anormal eğriliği (Görsel 3) de ortaya çıkabilir.

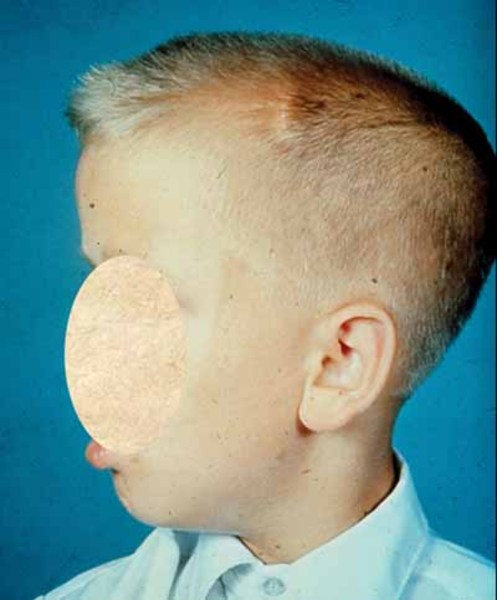
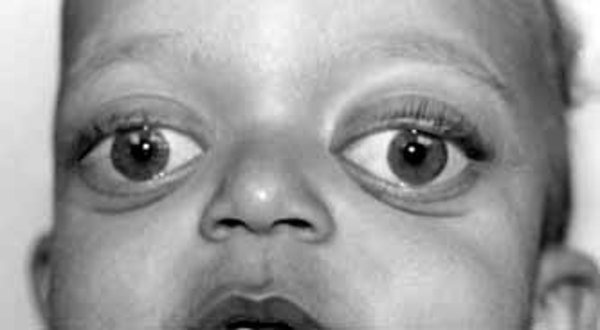
Camurati-Engelmann Hastalığı olan bireyler, anormal derecede büyük bir baş (makrosefali) (Görsel 4) ve alt çene (çene), belirgin bir alın (ön alın) (Görsel 5) ve sığ göz yuvaları (oküler proptoz) (Görsel 6) ile şişkin gözlere yol açabilen alışılmadık derecede kalın bir kafatasına sahip olabilir. Baş ve yüzdeki bu değişiklikler, yaş ile daha belirgin hale gelir ve etkilenen yetişkinlerde daha belirgindir. Camurati-Engelmann Hastalığı’na sahip bireylerin yaklaşık dörtte birinde, kalınlaşmış kafatası beyindeki basıncı arttırır ya da omuriliği sıkıştırır, bu da baş ağrısı, işitme kaybı, görüş problemleri, baş dönmesi (vertigo), kulak çınlaması (tinitus) ve yüz felci gibi çeşitli nörolojik problemlere sebep olabilir.

Hiperostoz derecesi, ilk semptomlarını yaşadığı Camurati-Engelmann Hastalığı olan bireyler arasında değişir.
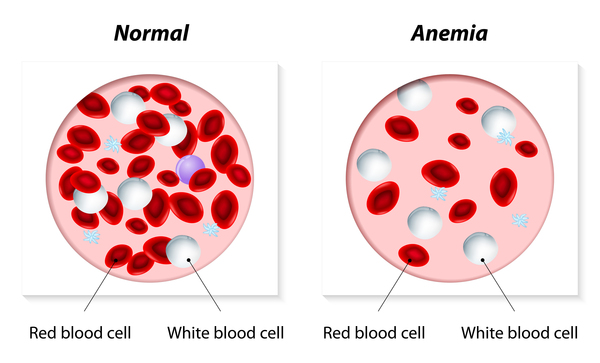
Camurati-Engelmann Hastalığı’nın diğer nadir özellikleri, boya oranla anormal derecede uzun uzuvlar, kas kütlesinde ve vücut yağında düşüş, gecikmiş diş çıkarma (dentisyon), sık boşluklar, gecikmiş ergenlik, kırmızı kan hücresi yetersizliği (anemi) (Görsel 7), genişlemiş karaciğer ve dalak (hepatosplenomegali), derinin incelmesi ve aşırı terli el ve ayakları (hiperhidrotik) içerir.
Sıklık
Camurati-Engelmann Hastalığı’nın yaygınlığı bilinmemektedir. Dünya çapında 300’den fazla vaka bildirilmiştir.
Nedenler
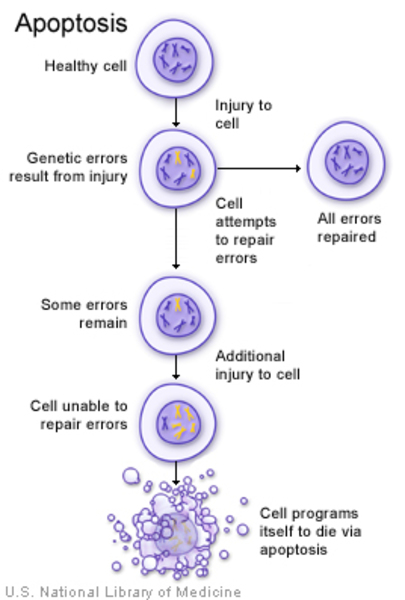
TGFB1 genindeki mutasyonlar Camurati-Engelmann Hastalığı’na neden olur. TGFB1 geni, transforme edici büyüme faktörü beta-1 (TGF)-1) adı verilen bir proteinin üretim emrini verir. TGFβ-1 proteini, hücrenin büyüme ve bölünmesi (çoğalması), belirli fonksiyonları yerine getirmesi için hücrenin olgunlaşması (farklılaşma), hücre hareketi (motilite) ve kontrollü hücre ölümü (apoptoz)(Görsel 8) dahil olmak üzere çeşitli hücre aktivitesini düzenleyen kimyasal sinyalleri tetikler.
TGFβ-1 proteini vücutta bulunur, ancak iskeleti oluşturan dokularda bol miktarda bulunur, burada erken gelişim sırasında iskeletin çoğunu oluşturan sert, esnek bir doku olan kemik ve kıkırdak oluşumunu ve büyümesini düzenlemeye yardımcı olur. TGFβ-1, diğer dokularda farklı işlemlerde yer almaktadır.
Camurati-Engelmann Hastalığı’na neden olan TGFB1 gen mutasyonları, aşırı aktif TGFβ-1 proteininin üretilmesine sebep olur. Bu anormal TGFβ-1 protein aktivitesi, daha fazla kemik oluşumuna sebep olan sinyallerde artışa sebep olur. Sonuç olarak, kollardaki, bacaklardaki ve kafatasındaki kemikler normalden daha kalındır ve Camurati-Engelmann Hastalığı’na sahip bireylerin sıkça yaşadığı hareket ve nörolojik problemlere katkıda bulunur.
Bazı Camurati-Engelmann Hastalığı’na sahip olan bireyler, TGFB1 geninde tanımlanmış bir mutasyona sahip değildir. Bu durumlarda, durumun nedeni bilinmemektedir.
Kalıtım Modeli
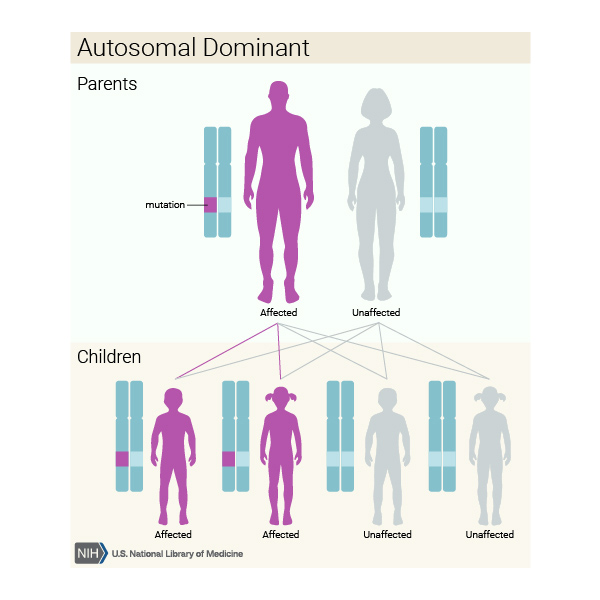
Bu durum otozomal dominant bir modelde kalıtsaldır, yani her hücredeki değiştirilmiş genin bir kopyasının bozukluğa neden olması için yeterli olduğu anlamına gelir.
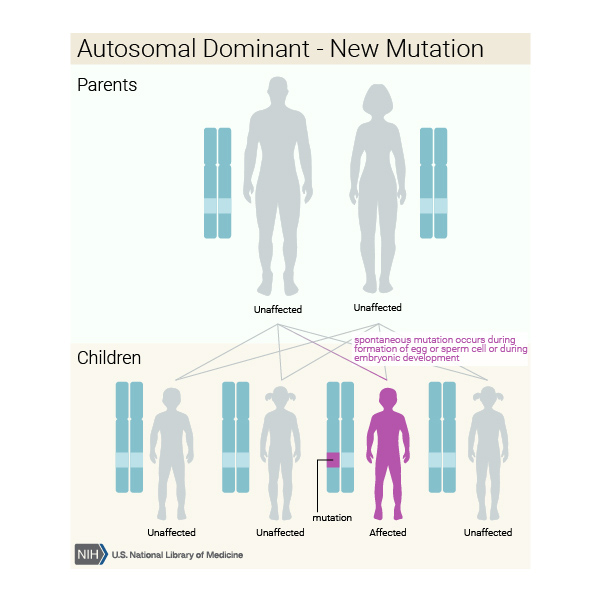
Bazı durumlarda, etkilenen bir kişi, etkilenen bir ebeveyninden (Görsel 9) mutasyonu devralır. Diğer vakalar, gendeki yeni mutasyonlardan (Görsel 10) kaynaklanır ve ailelerinde hastalık öyküsü olmayan kişilerde ortaya çıkar.
Değiştirilmiş gen sahibi olan bazı kişilerde, azalmış penetrasyon (etkililik) olarak bilinen durum asla gelişmez.
Bu Hastalığın Diğer İsimleri
- Camurati-Engelmann Sendromu
- CED
- Diyafiz Displazisi
- Diyafiz Hiperostozu
- Diyafiz Osteosklerozu
- Engelmann Hastalığı
- PDD
- İlerleyici Diyafiz Displazisi
Kaynak: https://ghr.nlm.nih.gov/condition/camurati-engelmann-disease#
Görsel Kaynak: https://www.spandidos-publications.com/10.3892/mmr.2013.1367
Editör: Meryem Melisa KAR
