
İçindekiler
ATP7A Geni: ATPase Bakır Taşıma Alfa
Normal Fonksiyon
ATP7A geni, vücuttaki bakır seviyelerini düzenlemek için önemli olan bir proteinin yapım emrini verir. Pek çok hücresel fonksiyon için bakır gereklidir ancak aşırı miktarda bulunduğunda toksiktir. ATP7A proteini, karaciğer hücreleri hariç vücutta bulunur. İnce bağırsakta bu protein, bakırın gıdalardan emilimini kontrol etmeye yardımcı olur. Diğer hücrelerde, ATP7A proteini ikili bir role sahiptir ve iki hücresel konum arasında mekik bulunur. Protein normal olarak, enzimler dahil olmak üzere yeni üretilen proteinleri değiştiren, golgi aparatı adı verilen bir hücre yapısında bulunur. Golgi aparatında, ATP7A proteini kemik, cilt, saç, kan damarları ve sinir sisteminin yapısı ve işlevi için kritik olan bazı enzimlere bakır sağlar. Bununla birlikte, hücre ortamındaki bakır seviyeleri yükselirse, ATP7A proteini hücre zarına hareket eder ve hücredeki fazla bakır miktarını ortadan kaldırır.
Genetik Değişikliklerle İlgili Sağlık Durumları
Kutis Laksa
ATP7A genindeki çeşitli mutasyonlar, Oksipital Horn Sendromu veya hafif bir Menkes Sendromu şekli olarak kabul edilen X’e bağlı Kutis Laksa denilen bir durumdan sorumludur. Oksipital Horn Sendromu, gevşek ve sarkan cilt, kafatasının tabanındaki bir kemikte kama şeklinde kalsiyum birikintileri (oksipital kemik), kaba saç ve gevşek eklemler ile karakterize edilmiştir.
Oksipital Horn Sendromu’na neden olan mutasyonların çoğu, ATP7A proteininin üretimini azaltır fakat ortadan kaldırmaz. Bu proteinin yetersizliği, bakırın gıdalardan emilimini engeller ve vücuttaki hücrelere normal dağılımını önler. Düşük bakır kaynağı, kemik, cilt, saç, kan damarlarının ve sinir sisteminin yapısını ve işlevini etkileyen sayısız bakır içeren enzimin aktivitesini azaltabilir. Bu enzimlerin azalmış aktivitesi, Oksipital Horn Sendromu’nun karakteristik özelliklerini oluşturur.
Menkes Sendromu
Araştırmacılar, Menkes Sendromu’na neden olan ATP7A geninde 150’den fazla mutasyon saptadılar. Bu mutasyonların çoğu genin bir kısmını siler ve muhtemelen kısaltılmış bir ATP7A proteini ile sonuçlanır. Diğer mutasyonlar gene ek olarak DNA yapıtaşları (nükleotitler) ekler veya tek nükleotidleri değiştirir. Bu mutasyonların tümü, fonksiyonel ATP7A proteini üretimini önler. Sonuç olarak, bakırın gıdadan emilimi bozulur ve bazı enzimlere bakır verilmez. Anormal protein hücre zarına sıkışabilir ve golgi aparatından ileri geri hareket edemeyebilir.
ATP7A proteininin bozulan etkinliği, bakırın vücuttaki hücrelere zayıf şekilde dağılmasına neden olur. Bakır, ince bağırsak ve böbrekler gibi bazı dokularda birikirken, beyin ve diğer dokular alışılmadık derecede düşük seviyelere sahiptir. Düşük bakır kaynağı, kemik, cilt, saç, kan damarlarının ve sinir sisteminin yapısını ve işlevini etkileyen bakır içeren birçok enzimin aktivitesini azaltabilir. Menkes Sendromu’nun belirtileri ve semptomları, bu bakır içeren enzimlerin aktivitesinin azalmasından kaynaklanır.
Charcot-Marie-Tooth Hastalığı
Kromozomal Konum
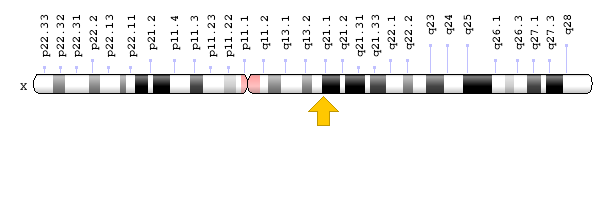
- Sitogenetik Konum: Xq21.1, 21.1 konumundaki X kromozomunun uzun (q) kolu
- Moleküler Konum: X kromozomu üzerinde 77,910,656 – 78,050,395 baz çiftleri
Bu Gen İçin Diğer İsimler
- ATP7A_İNSAN
- ATPase, Cu ++ Taşıma, Alfa Polipeptit
- ATPase, Cu ++ Taşıma, Alfa Polipeptit (Menkes Sendromu)
- ATPP1
- Bakır Pompa 1
- MC1
- MK
- MNK
- İSG
Kaynak: https://ghr.nlm.nih.gov/gene/ATP7A
Görsel Kaynak: https://www.rcsb.org/3d-view/ngl/2kij
Editör: Meryem Melisa KAR
